
ولتامتری،ولتاموگرام
در این مقاله توضیحاتی در مورد ولتامتری و قرارداد های رسم ولتاموگرام و اندازه گیری جریان محدودکننده داده خواهد شد.
ولتامتری چیست
اصطلاح ولتامتری به طور گسترده به هر روشی اشاره دارد که در آن پتانسیل الکترود در حین اندازهگیری جریان تغییر میکند. اصطلاحات مرتبط با ولتامتری در صنایع مختلف و رشته های دانشگاهی متفاوت است، اما اصول اساسی همه تکنیک های ولتامتری بسیار مشابه هستند.
ولتامتری
مفاهیم پایه الکتروشیمیایی در دورههای شیمی دبیرستان به دانشجویان آموزش داده میشود و آن ها قبلاً با نیمه واکنش ها، پتانسیل های استاندارد، الکترودهای مرجع، معادله نرنست، محاسبات انرژی آزاد و سلول های الکترود دوگانه ساده آشنا شده اند(برای مطالعه بیشتر کلیک کنید.).
بیشتر محاسباتی که دانشجویان در این دوره ها انجام داده اند با موضوع پتانسیومتری سروکار دارد.
یعنی داده های استوکیومتری و غلظتی که سلول الکتروشیمیایی را توصیف می کند به آنها داده می شود و از آنها خواسته می شود پتانسیل سلول را پیش بینی کنند.
این نوع مطالب به دانجشویان دید بسیار محدودی از کاربرد الکتروشیمی میدهد دانشآموزان کاملاً گیج میشوند و بیش از حد نگران این هستند که چگونه بفهمند کدام الکترود کاتد است یا کدام نیمهواکنش «برگشت پذیر» میشود.
شما میتوانید با کمک مقاله بررسی سه روش ولتامتری و ولتامتری چرخه ای: تعین غلظت آهن مجهول آزمایشاتی کاربردی برای دانشجویان تعریف کنید.
قدرت تکنیکهای الکتروشیمیایی مدرن از یک ایده بسیار ساده ناشی میشود:
- فرآیند ردوکسی که در یک سلول الکتروشیمیایی انجام می شود، انتقال الکترون بین واکنشدهندهها را از یک مدار الکترونیکی خارجی عبور می دهد.
از آنجایی که طراحی و کنترل مدار خارجی در دست الکتروشیمیدان است، جریان الکترون ممکن است به هر طریقی که مورد نظر باشد، مانند موارد زیر استفاده شود.
-
منبع نیرو،
-
آنالیزشیمیایی
-
انجام واکنش الکتروسنتز
الکتروشیمیدان مدرن به جای مشاهده غیرفعال فرآیند ردوکس، کنترل فرآیند را به دست میگیرد و :
-
سرعت وقوع واکنش (با تنظیم جریان)
-
میزان وقوع واکنش (با تنظیم پتانسیل)
را تنظیم میکند.
همچنین باید تاکید کرد که جریان و پتانسیل را نمی توان به طور همزمان کنترل کرد و الکتروشیمیدان باید انتخاب کند که کدام متغیر آزمایشی باید کنترل شود.
این راهنمای آزمایشگاهی منحصراً به ولتامتری می پردازد، که به تکنیک هایی اشاره دارد که در آن پتانسیل یک الکترود کنترل شده است و جریان متغیر مشاهده می گردد. تکنیک های برعکس آن، روش های گالوانوستاتیک نامیده می شوند.
برای کمک به درک درست دانش آموز از ولتامتری، با مقایسه آن با پتانسیومتری شروع می کنیم. همچنین چند مفهوم اساسی باید مورد بحث قرار گیرد تا به پیوند این دو کمک کند و احتمالاً بحث در مورد معادله نرنست و قانون فارادی در هر زمینه کافی است.
سپس، دانشجویان به آرامی تشویق میشوند که نگران اتفاقی که در هر الکترودِ غوطهور در محلول آزمایشی میافتد، نباشد، بلکه روی آنچه که فقط در یک الکترود، یعنی الکترود کار میافتد، تمرکز کند.
پاراگراف های زیر باید یک طرح کلی مفید از نحوه انتقال از پتانسیومتری به ولتامتری ارائه دهند.
تفاوت پتانسیومتری با ولتامتری
معادله نرنست رابطه بین پتانسیل الکترود و غلظت محلول را توصیف می کند.
در کاربرد آشنا (و نسبتاً خسته کننده) پتانسیومتری، الکتروشیمیدان نقش منفعلی دارد. این بدان معناست که غلظت محلول هر چه باشد هست، و الکتروشیمیدان فقط پتانسیل الکترود کار را اندازه میگیرد، که در تئوری، همان چیزی است که توسط معادله نرنست پیشبینی میشود.
در زمینه هیجان انگیزتر ولتامتری، همه چیز برعکس است و الکتروشیمیدان نقش فعالی را بر عهده می گیرد. الکتروشیمیدان با کنترل پتانسیل الکترود، غلظت محلول در مجاورت الکترود را تحت تأثیر قرار می دهد.
البته، معادله نرنست همچنان برای پیشبینی غلظت گونههای نزدیک به سطح الکترود در یک پتانسیل کاربردی معین مفید است. با این حال، تنها در مواردی که سیستم ردوکس به سرعت به تغییرات پتانسیل اعمال شده پاسخ دهد، تصویر دقیقی ارائه می دهد.
چنین سیستمهای ردوکس با سینتیک سریع، در شبه تعادل با سطح الکترود باقی میمانند و به همین دلیل گاهی اوقات سیستمهای «نرنستین» یا برگشتپذیر نامیده میشوند.
پتانسیل الکترود کار تعیین می کند که چه فرآیندهای ردوکسی در سطح الکترود (در صورت وجود) رخ می دهد.
-
پتانسیل های بسیار مثبت احتمالاً آنالیت ها را اکسید می کند
-
پتانسیل های منفی احتمالاً گونه ها را کاهش می دهد.
در هر صورت، جریانی از الکترود عبور میکند و مقدار این جریان معمولاً متناسب با غلظت محلول است.
بنابراین، جریان، سیگنال تحلیلی را می دهد که می تواند به عنوان مبنایی برای منحنی کالیبراسیون مربوط به سیگنال به غلظت استفاده شود.
سیستم های الکتروشیمیایی لزوما ناهمگن هستند:
تمام رویدادهای شیمیایی و الکتروشیمیایی در سطح یا نزدیک سطح الکترود کار رخ می دهد.
این بدان معنی است که بخشی از محلولِ بسیار نزدیک به الکترود، به نوعی خاص است زیرا تحت تأثیر آن قرار می گیرد.
به طور کلی، الکترود بر لایه ای از محلول تأثیر می گذارد که فقط چند میکرومتر از سطح الکترود فاصله دارد.
غلظت آنالیت در این لایه انتشار بسیار کوچک، می تواند به طور گسترده ای با غلظت های موجود در بخش عمده محلول متفاوت باشد.
در مقایسه با حجم کلی محلول در سلول، حجم لایه انتشار ناچیز است و تغییرات غلظت ناشی از الکترود در لایه انتشار به ندرت بر محلول حجیم تأثیر می گذارد.
قانون فارادی بیان میکند که مقدار بار درگیر در یک فرآیند الکتروشیمیایی به طور مستقیم با استوکیومتری نیمهواکنش مرتبط است.
از آنجایی که اکثر نیمه واکنش ها شامل انتقال یک الکترون ساده است، استفاده از قانون فارادی به این بیانیه منجر می شود که به ازای هر مول آنالیت که در یک الکترود اکسید یا احیا می شود، یک مول الکترون با بار 96485 کولن، وارد می شود یا الکترود را ترک می کند.
برای آن نیمهواکنشهایی که شامل دو، سه یا چهار الکترون است، مقدار C/mole 96485 به سادگی در عدد صحیح مناسب ضرب میشود.
قانون فارادی ارتباط بین جریان مشاهده شده و غلظت آنالیت را فراهم می کند.
به طور کلی، غلظت های بالاتر به معنای جریان بیشتر است.
هنگامی که پتانسیل الکترود به اندازه کافی از مقدار تعادل دور شود، جریان تنها با سرعت رسیدن آنالیت به سطح الکترود محدود می شود.
در محلول های هم زده نشده، آنالیت تنها با انتشار، در سراسر لایه انتشار از محلول توده به سطح الکترود، می تواند به الکترود برسد.
سرعت انتشار توسط یک گرادیان غلظت کنترل می شود . یعنی انتشار از ناحیه ای با غلظت بالا به ناحیه ای با غلظت کم رخ می دهد و هر چه اختلاف غلظت بیشتر باشد، انتشار سریعتر اتفاق می افتد.(دانشجویان معمولاً از قبل با این ایدهها از مطالعه رفتار گاز ایدهآل آشنا هستند.)
این بدان معناست که جریان در یک الکترود زمانی بزرگتر است که گرادیان غلظت آنالیت (در سراسر لایه انتشار) بیشتر باشد.
در نهایت، باید به دانشآموز گفته شود که وقتی قانون فارادی با قانون اول انتشار فیک ترکیب میشود، یک عبارت کلی برای جریان محدود انتشار، id، در یک الکترود کار به دست میآید:
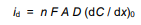
که در آن
- n تعداد الکترون های درگیر در نیمه واکنش،
- F ثابت فارادی،
- A ناحیه الکترود،
- D ضریب انتشار آنالیت و
- 0(dC/dx) گرادیان غلظت در سطح الکترود است.
این عبارت بر خلاف معادله نرنست برای دانش آموز جدید خواهد بود و بسیار مهم است که بر رابطه متناسب بین گرادیان جریان و غلظت تاکید شود.
به یک معنا، تمام تکنیکهای ولتامتری محدود با انتشار:
-
ولتامتری چرخهای،
-
ولتامتری دیسک چرخشی،
-
کرونوآمپرومتری،
و غیره توسط معادله بالا کنترل میشوند.
این می تواند یک تقسیم بندی خوب برای دانشجوینیکه در مورد بیش از یک نوع ولتامتری یاد می گیرند باشد.
هر تکنیک تنها در شیوه ای که گرادیان غلظت (dC/dx)0 ایجاد و حفظ می شود متفاوت است. این چیزی است که به هر تکنیک پاسخ فعلی منحصر به فرد خود را می دهد.
انواع ولتامتری
مقاله انواع ولتامتری
- (شما میتوانید تکنیکهای مختلف را با دستگاه انالایزر الکتروشیمی BHP-2066 اجرا کنید.)
ولتامتری روبش خطی
رایج ترین شکل ولتامتری شامل روبش پتانسیل الکترود از مقدار اولیه به مقدار نهایی با سرعت ثابت است.
هنگام کار در زمینه شیمی الکتروتحلیلی با یک الکترود غیر دوار، این تکنیک ولتامتری روبش خطی (LSV) نامیده می شود. در زمینه علم خوردگی، این نوع تکنیک معمولاً مقاومت قطبش خطی (LPR) یا آنالیز تافل نامیده می شود.
اصطلاح ولتامتری چرخه ای (CV) به روشی اشاره دارد که در آن پتانسیل الکترود به طور مکرر بین دو پتانسیل به جلو و عقب جابجا می شود.
در کار با الکترود دوار، روبش پتانسیل معمولاً حداقل 200 میلی ولت در دو طرف پتانسیل الکترود استاندارد را در بر می گیرد و نرخ چرخش معمولاً بین 100 تا 2400 RPM است.(سرعت چرخش ثابت نگه داشته میشود، زیرا الکترود از یک پتانسیل به پتانسیل دیگر با سرعت جابجایی ثابت روبش میشود.)
با این حال، در زمینه یک مطالعه خوردگی، روبش پتانسیل ممکن است محدوده بسیار باریک تری (50 میلی ولت) را با استفاده از سرعت روبش آهسته تر (کمتر از 5 میلی ولت در ثانیه) با تاکید بر نرخ های چرخش بالاتر داشته باشد.
به عنوان مثال، محلولی را در نظر بگیرید که در ابتدا فقط دارای شکل اکسید شده یک مولکول یا یون است.
یک الکترود در این محلول ابتدا در پتانسیل مثبت تر از پتانسیل استاندارد قرار می گیرد.
- در این پتانسیل، جریان کمی وجود دارد یا اصلاً وجود ندارد، زیرا چیزی برای اکسید شدن وجود ندارد (مولکول یا یون قبلاً اکسید شده است)، و پتانسیل آنقدر منفی نیست که باعث کاهش محسوس مولکول یا یون شود.
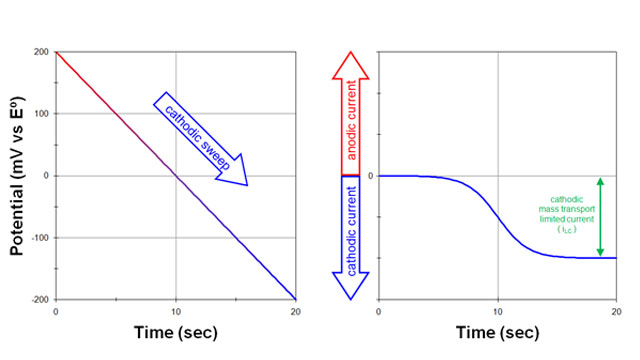
بعد، پتانسیل الکترود به آرامی (20 میلی ولت در ثانیه) در جهت منفی (کاتدی) روبش می شود (شکل 1. سمت چپ).
- همانطور که پتانسیل اعمال شده به پتانسیل الکترود استاندارد نزدیک می شود، یک جریان کاتدی مشاهده می شود (شکل 1. سمت راست). با عبور از پتانسیل الکترود استاندارد به سمت پتانسیل های منفی تر، جریان کاتدی به افزایش خود ادامه می دهد.
هنگامی که پتانسیل اعمال شده به اندازه کافی نسبت به پتانسیل الکترود استاندارد منفی شود:
- جریان در نهایت به حداکثر مقدار (جریان محدود کننده) می رسد. در چنین پتانسیل منفی، هر گونه اکسید شده مولکول یا یون (O) که به سطح الکترود می رسد بلافاصله به شکل کاهش یافته (R) تبدیل می شود.
O+ne–>R
جریان کاتدی مشاهدهشده نتیجه جریان الکترونهایی است که از الکترود خارج میشوند و به داخل محلول میروند. سرعت جریان الکترون تنها با سرعتی که شکل اکسید شده (O) میتواند به سطح الکترود برسد محدود میشود. حداکثر جریان مشاهده شده در این شرایط، جریان محدود کننده کاتدی (iLC) نامیده می شود.
هرگاه یک جریان مشاهده شده فقط با سرعتی که مواد به سطح الکترود می رسد محدود شود، گفته می شود که جریان محدود به انتقال جرم است.
اما هنگام کار با یک الکترود دوار، سرعت انتقال جرم با سرعت چرخش الکترود مرتبط است. چرخش الکترود با سرعت بیشتر، سرعت رسیدن مواد به سطح الکترود را افزایش می دهد. بنابراین، جریان محدود کننده با افزایش سرعت چرخش افزایش می یابد. آزمایشهای مربوط به یک الکترود دوار برای بهرهبرداری هدفمند از این رابطه اساسی بین سرعت چرخش و جریان محدودکننده طراحی شدهاند.
آزمایش روبش کاتدی که در بالا توضیح داده شد (شکل 1) در موردی اعمال می شود که محلول در ابتدا فقط حاوی فرم اکسید شده O مولکول یا یون مورد مطالعه باشد. حالت مخالف نتایج مشابهی را به همراه دارد.
محلولی را در نظر بگیرید که در ابتدا فقط دارای شکل احیا شده R از مولکول یا یون مورد مطالعه است.
الکترود دوار در ابتدا در پتانسیلی قرار می گیرد که حدود 200 میلی ولت منفی تر از پتانسیل استاندارد است.
- در این پتانسیل، جریان کمی وجود دارد یا اصلاً وجود ندارد زیرا چیزی برای کاهش وجود ندارد (مولکول یا یون قبلاً کاهش یافته است) و پتانسیل آنقدر مثبت نیست که باعث اکسیداسیون قابل ملاحظه مولکول یا یون شود.
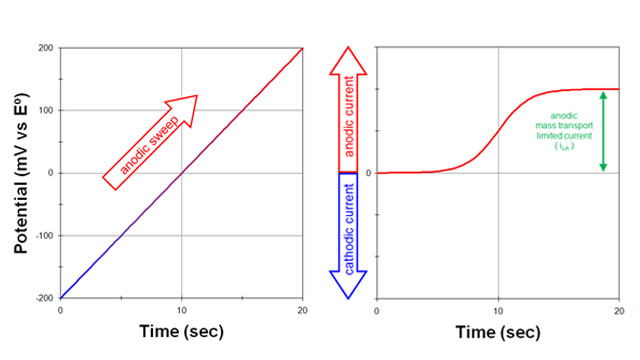
سپس، پتانسیل الکترود به آرامی در جهت مثبت (آندی) روبش می شود (شکل 2. سمت چپ):
- یک جریان آندی مشاهده می شود (شکل 2. سمت راست).
جریان آندی در نهایت زمانی به حداکثر مقدار می رسد که پتانسیل نسبت به پتانسیل الکترود استاندارد به اندازه کافی مثبت باشد.
- در این مرحله، هر یک از فرم احیا شده (R) که به سطح الکترود می رسد بلافاصله به فرم اکسید شده (O) تبدیل می شود.
R–> O+ne
جریان مشاهده شده نتیجه جریان الکترون ها به داخل الکترود است. حداکثر جریان مشاهده شده جریان محدود کننده آندی (iLA) نامیده می شود.
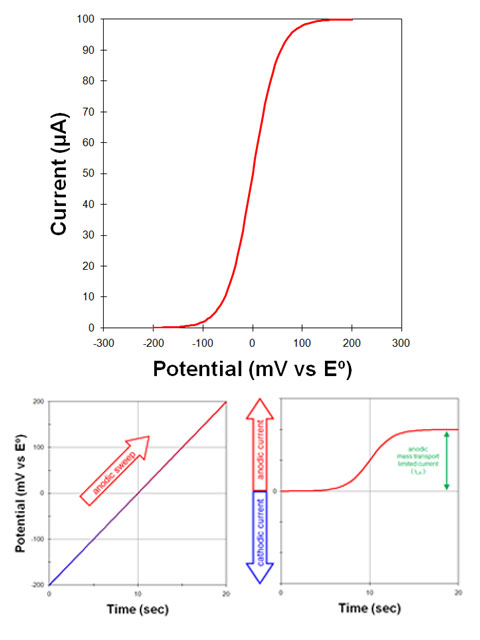
قراردادهای ترسیم ولتاموگرام voltamogram
دو جریان داده ثبت شده در طول آزمایش ولتامتری عبارتند از:
-
پتانسیل در مقابل زمان
-
جریان در برابر زمان.
به جای رسم مجزای این دو جریان (شکل 3. پایین به چپ)، ترسیم جریان در مقابل پتانسیل (شکل 3. بالا) رایج تر است. چنین نموداری ولتاموگرام نامیده می شود.
اگرچه اکثر محققان الکتروتحلیلی موافق هستند که جریان باید در امتداد محور عمودی و پتانسیل در امتداد محور افقی ترسیم شود، هیچ توافق گسترده ای در مورد جهت (جهت) هر محور وجود ندارد.
- برخی از محققان پتانسیل های مثبت (آندی، اکسید کننده) را به سمت راست ترسیم می کنند
- برخی پتانسیل منفی (کاتدی، کاهنده) را به سمت راست ترسیم می کنند (طبق سنت پلاروگرافی کلاسیک).
علاوه بر این،
- برخی از محققان جریان آندی (اکسید کننده) را به سمت بالا در امتداد محور عمودی ترسیم می کنند،
- برخی دیگر جریان کاتدی (کاهنده) را در جهت بالا ترسیم می کنند.
این بدان معناست که چهار قرارداد ممکن برای ترسیم یک ولتاموگرام وجود دارد، و همیشه باید قبل از تفسیر ولتاموگرام، جهت گیری محورها را مشخص کرد.
خوشبختانه، از چهار راه ممکن برای رسم ولتاموگرام، تنها دو روش معمولا استفاده می شود.
- سنت قدیمی (بر اساس پلاروگرافی کلاسیک) جریان کاتدی را به سمت بالا در امتداد محور عمودی و پتانسیل های منفی (کاتدی، کاهنده) را به سمت راست در امتداد محور افقی ترسیم می کند.
یک ولتاموگرام پیچیده شامل چهار جریان محدود کننده مختلف (شکل 4. سمت چپ) این قرارداد را نشان می دهد که گاهی اوقات قرارداد “آمریکای شمالی” نامیده می شود.
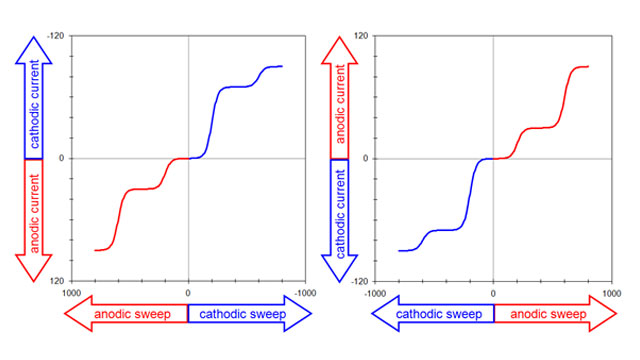
همان داده ها را می توان با استفاده از قرارداد “اروپایی” ترسیم کرد (شکل 4. سمت راست).
- این قرارداد جریان های آندی را به سمت بالا در امتداد محور عمودی و پتانسیل های مثبت (آندی، اکسید کننده) بیشتر را به سمت راست در امتداد محور افقی ترسیم می کند.
قرداد اروپایی توسط کسانی که خارج از جامعه تحقیقاتی الکتروتحلیلی هستند به راحتی قابل درک است (زیرا مقادیر مثبت به سمت راست در امتداد محور افقی ترسیم می شوند).
قرداد اروپایی در بقیه این سند استفاده می شود. توجه داشته باشید که این انتخاب همچنین مستلزم یک قرارداد علامت ریاضی برای جریان است. به طور خاص، مقادیر جریان مثبت، آندی و مقادیر جریان منفی در این سند کاتدی در نظر گرفته می شوند.
این قرارداد علامت تا حدودی دلخواه است و نرم افزار پردازش داده های الکتروشیمیایی موجود از تولید کنندگان مختلف ممکن است از این قرارداد علامت استفاده کند یا نکند.(نرم افزار BHP مختص آنالایزر الکتروشیمیایی شرکت به پژوه از این قرارداد استفاده می کند.)
اندازه گیری جریان های محدود کننده
پاسخ ولتامتری نظری از یک الکترود یک موج سیگموئیدی متقارن است (مانند ولتاموگرام های ایده آل نشان داده شده در شکل 3 و شکل 4). یک سیگموئید کامل دارای یک جریان زمینه صاف قبل از موج و یک جریان محدود کننده صاف بعد از موج است.
ارتفاع پیک (که از جریان پایه (baseline) تا خط تراز (plateau) جریان محدودکننده، اندازه گیری می شود) جریان محدودکننده، جریان انتقال جرم است.
در آزمایشهای واقعی پیک ممکن است در بالای جریان پسزمینه مشاهده شود، و علاوه بر این، جریان پسزمینه ممکن است کمی شیب داشته باشد (شکل 5).
این جریان پس زمینه (ناخواسته) ممکن است به دلایل زیر باشد :
-
تداخل اکسیداسیون ، کاهش ناخالصی ها یا حلال و
-
شارژ و تخلیه خازنی لایه دوگانه یونی که در کنار سطح الکترود پلاریزه تشکیل می شود.
هنگام تلاش برای اندازهگیری جریان محدود انتقال جرم فارادیک در یک الکترود دوار، اغلب لازم است که جریان پسزمینه در نظر گرفته شود.
اگر جریان پسزمینه شیب ثابتی در کل ولتاموگرام داشته باشد، برونیابی خط مبنای شیبدار به نقطهای زیر خط تراز جریان محدودکننده نسبتاً آسان است (شکل 5).
- جریان محدود کننده به عنوان فاصله (عمودی) بین خط تراز و خط پایه برون یابی اندازه گیری می شود. در ولتاموگرام هایی که بیش از یک موج وجود دارد، خط تراز برای موج اول به عنوان خط پایه برای موج دوم استفاده می شود (شکل 5. سمت چپ).
در برخی موارد، شیب جریان پسزمینه در کل ولتاموگرام ثابت نیست. یعنی شیب خط پایه منتهی به موج می تواند با شیب خط تراز بعد از موج متفاوت باشد. تشخیص دقیق محل اندازه گیری جریان محدود کننده در طول چنین ولتاموگرام بسیار دشوار است.
- یک روش برون یابی خط مبنا به جلو و برون یابی خط تراز به عقب است. سپس، جریان محدود کننده به عنوان فاصله عمودی بین خط مبنا و خط تراز در نقطه ای مطابق با مرکز ولتاموگرام اندازه گیری می شود (شکل 5. سمت راست).
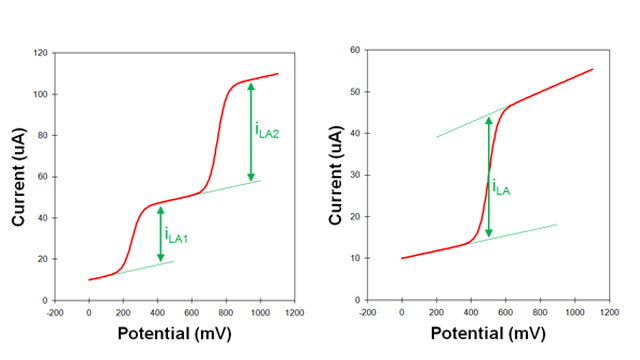
در نرم افزار BHP مختص انالایزر الکتروشیمیایی شرکت به پژوه با استفاده از قابلیت پیک سرچ میتوان جریان پیک را محاسبه کرد.
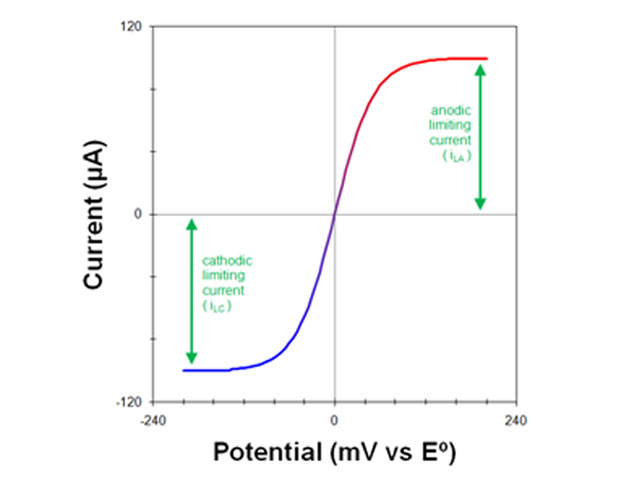
در نهایت، باید توجه داشت که وقتی شکل اکسید شده O و شکل احیا شده R یک مولکول یا یون هر دو در یک محلول به طور همزمان وجود داشته باشند، ولتاموگرام احتمالاً یک جریان محدود کننده کاتدی و آندی را نشان می دهد (شکل 6).
اندازه گیری درست جریان محدود کننده در این مورد می تواند بسیار دشوار باشد، به خصوص اگر یک جریان پس زمینه شیب دار نیز وجود داشته باشد. به همین دلیل، بیشتر آزمایشها با الکترودهای دوار در محلولهایی انجام میشوند که در ابتدا فقط یک شکل از مولکول یا یون وجود دارد.
voltametery رفرنس
(1) Kissinger, P.; Heineman, W. R. Laboratory Techniques in Electroanalytical Chemistry; 2nd ed.;
Marcel Dekker, Inc: New York, 1996.
(2) Bard, A. J.; Faulkner, L. R. Electrochemical Methods – Fundamentals and Applications; 2nd ed.;
John Wiley {&} Sons: New York, 2000.
(3) Johnson, D. C.; Weber, S. G.; Bond, A. M.; Wightman, R. M.; Shoup, R. E.; Krull, I. S.
Electroanalytical Voltammetry in Flowing Solutions. Anal. Chim. Acta 1986, 180, 187–250.
(4) Gunasingham, H.; Fleet, B. Wall-Jet Electrode in Continuous Monitoring Voltammetry. Anal.
Chem. 1983, 55, 1409–1414.
(5) Macpherson, J. V; Unwin, P. R. Hydrodynamic Modulation Voltammetry with an Oscillating
Microjet Electrode. Anal. Chem. 1999, 71, 4642–4648.
(6) Henley, I. E.; Yunus, K.; Fisher, A. C. Voltammetry under Microfluidic Control: Computer-Aided
Design Development and Application of Novel Microelectrochemical Reactors. J. Phys. Chem.
B 2003, 107, 3878–3884.
(7) Pratt, K. W.; Johnson, D. C. Vibrating Wire Electrodes-I. Literature Review, Design and Evaluation.
Electrochim. Acta 1982, 27, 1013–1021.
(8) Hagan, C.; Coury, L. a. Comparison of Hydrodynamic Voltammetry Implemented by Sonication
to a Rotating Disk Electrode. Anal. Chem. 1994, 66, 399–405.
(9) Levich, V. G. Physicochemical Hydrodynamics; 1st ed.; Prentice-Hall: Englewood Cliffs, NJ, 1962.